Hurtige fakta
Find hurtige svar på spørgsmål, du måtte have om cystinose
Hos mennesker med cystinose opbygges cystin (en aminosyre) i en del af cellen kaldet lysosomet, og cellerne er ikke i stand til at fjerne det. Cystinose er en arvelig sygdom, der overføres fra forælder til barn, når en abnormitet i CTNS-genet fører til problemer med den måde, hvorpå cystin opbevares i kroppen. Cystinose kan kun udvikle sig hos børn, der modtager en ikke-fungerende kopi af cystinose-genet fra hver forælder.1
Der er tre typer af cystinose, som er klassificeret efter, hvor alvorligt nyrerne er påvirket, og i hvilken alder symptomerne begynder at vise sig; infantil nefropatisk cystinose (den mest almindelige form), juvenil eller sent opstået cystinose og voksen/okulær cystinose.1
Fordi nefropatisk cystinose påvirker hver celle i kroppen, er dens symptomer meget brede og forskellige. Selvom nyrerne først påvirkes, risikerer næsten alle organer i kroppen at blive beskadiget.2
Patienter med nefropatisk cystinose virker normale ved fødslen, men inden for det første leveår viser de ofte tegn, der tyder på, at deres nyrer ikke fungerer så godt, som de burde. Patienter kan også have nedsat vækst, appetitløshed eller rakitis.2
For alle oplysninger om symptomerne på cystinose gennem forskellige stadier af livet, henvises der til siden Symptomer.
Cystinose er en arvelig sygdom, der overføres fra forælder til barn, når et gen (CTNS-gen), der ikke fungerer, som det skal, fører til problemer med den måde, hvorpå cystin opbevares i kroppen.1
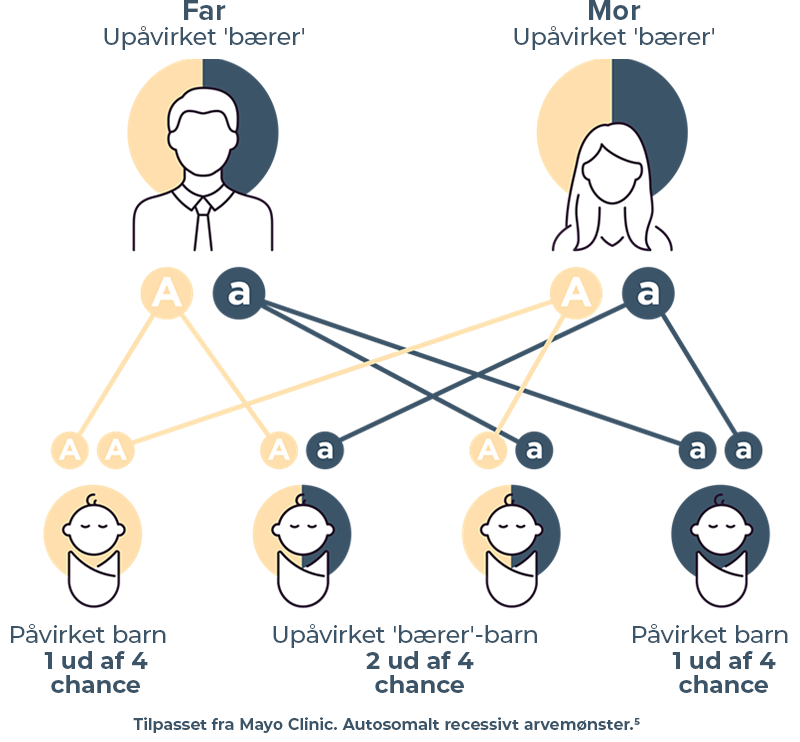
Cystinose kan kun udvikle sig hos børn, der modtager en ikke-fungerende kopi af cystinose-genet fra hver forælder.3,4 For hver graviditet, hvor begge forældre er upåvirkede bærere:
• Der er en 1 ud af 4 chancer for, at begge forældre vil videregive en ikke-fungerende kopi af genet, og barnet vil have lidelsen.3,4
• Der er en 2 ud af 4 chancer for, at den ene forælder vil videregive genændringen, og den anden ikke vil, hvilket betyder, at barnet vil være bærer af lidelsen, men ikke have det.3,4
• Der er en 1 ud af 4 chancer for, at begge forældre vil videregive en arbejdskopi af genet, og barnet ikke vil have lidelsen og ikke være bærer.3,4
Cystinose er en ultrasjælden sygdom, der rammer færre end 2 personer ud af hver million.2
Cystinose kan diagnosticeres ved hjælp af en test, der ser på niveauet af cystin i de hvide blodlegemer.1 Dette udføres normalt i et ekspertcenter af en nyrespecialist (pædiatrisk nefrolog) eller stofskiftespecialist.2 En spaltelampeundersøgelse kan også udføres af en øjenlæge for at se efter øjenkrystaller, som dukker op ved 1–2 års alderen.2 Hvis det er muligt, kan genetisk testning for genmutationer også udføres af en genetiker for at bekræfte diagnosen og karakterisere den specifikke mutation, som en patient har.1
Ved 6 til 12 måneder omfatter symptomer på cystinose: Fanconi-syndrom, overdreven urinproduktion (polyuri), overdreven tørst (polydipsi), dehydrering, tab af elektrolytter i urinen, glukose i urinen (glucosuri) og normale blodsukkerniveauer, aminosyrer til stede i urinen (aminoaciduri), væksthæmning og rakitis.1,2
Efter 1 til 2 år omfatter symptomerne cystinkrystaller, der er til stede i øjet (ophobning af hornhindekrystal).1
Efter 3 til 4 år omfatter symptomerne lysfølsomhed (fotofobi).1
En vellykket håndtering af denne tilstand kræver en bred vifte af behandlinger. Primært er dette cystin-nedbrydningsterapi, som virker ved at reducere cystinniveauet i celler i hele kroppen. Cystin-nedbrydningsterapi gives både oralt og topisk (som øjendråber). Det kan være nødvendigt med nyretransplantation, som skal efterfølges af behandling, som dæmper immunsystemet. Andre behandlinger kan omfatte insulin til behandling af diabetes og thyroxin til at erstatte manglende skjoldbruskkirtelhormoner.2,6,7